Estimating the genotype error probability,
A simple example:
cases <- sample_data_Hp_w(n = 1000, w = 0.1, p = c(0.25, 0.25, 0.5))
tab <- table(cases$xT, cases$xR)
tab
#>
#> 0 1 2
#> 0 180 53 7
#> 1 55 193 99
#> 2 8 78 327Maximum likelihood estimation
w_mle <- wgsLR::estimate_w(tab)
w_mle
#> [1] 0.0952511
w_mle_se <- wgsLR::estimate_w_se(tab, w_mle)
w_mle_se
#> [1] 0.005846342The last lines estimate the standard error that will be used below to construct an approximate confidence interval.
Please be aware of the “Cautionary note” in the README (about not just using standard VCF-files).
Bayesian inference
The package contains a simple Metropolis-Hastings sampler (also see the “Using Stan for Bayesian inference” vignette):
w_b <- wgsLR::estimate_w_bayesian(tab) |> posterior_samples()
w_b_q <- quantile(w_b, c(0.025, 0.975))
hist(w_b, main = NULL, xlab = expression(hat(w)))
abline(v = mean(w_b), col = "deeppink3", lwd = 3)
abline(v = w_b_q[1], col = "deeppink3", lty = 2)
abline(v = w_b_q[2], col = "deeppink3", lty = 2)
abline(v = w_mle, col = "yellow3", lwd = 3)
abline(v = w_mle - 2*w_mle_se, col = "yellow3", lty = 2)
abline(v = w_mle + 2*w_mle_se, col = "yellow3", lty = 2)
legend("topright", col = c("deeppink3", "yellow3"), legend = c("Bayesian", "MLE"), lty = 1, lwd = 2)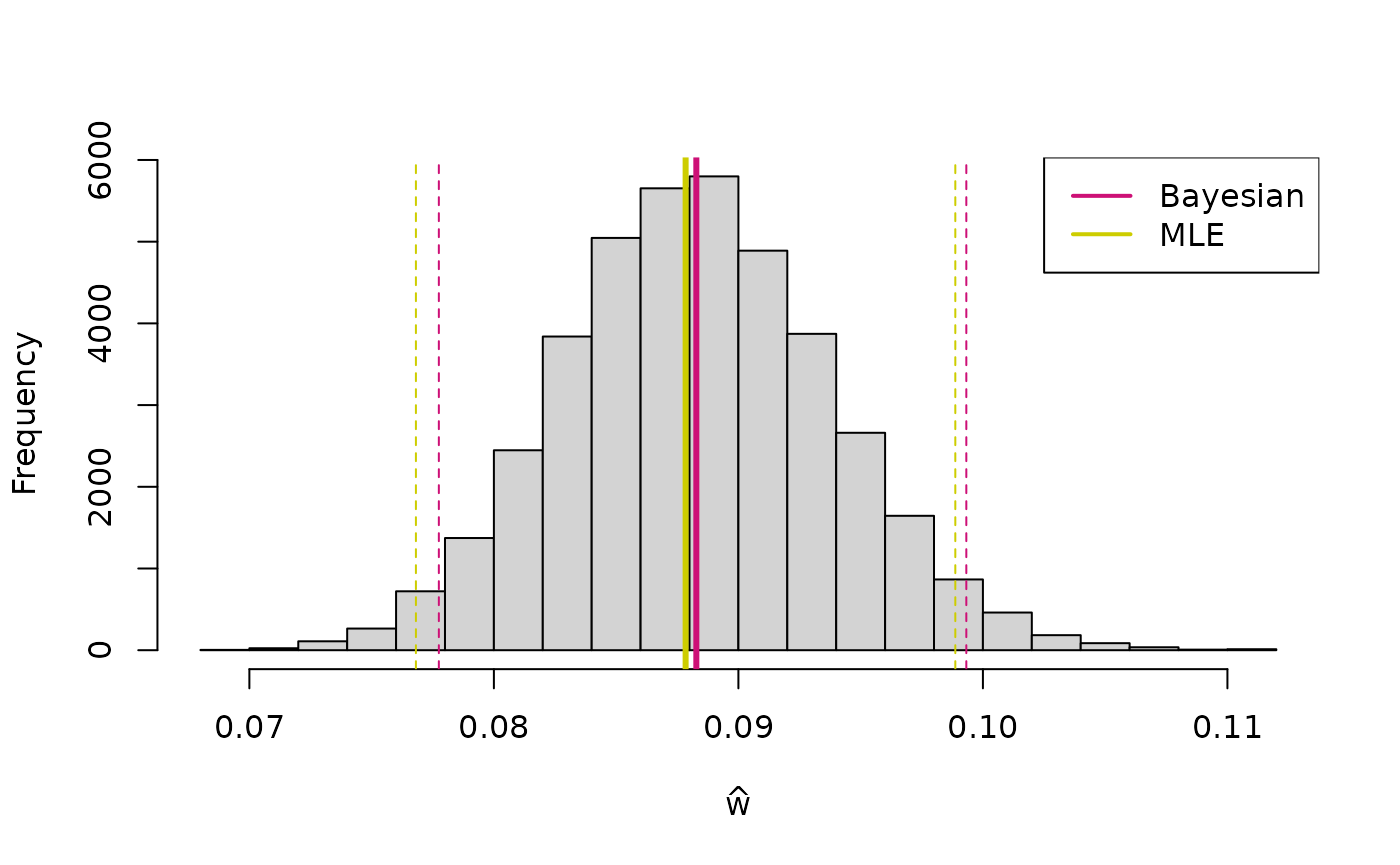
The dashed lines indicate approximate confidence intervals (maximum likelihood estimation) and credible interval (Bayesian inference).
Please be aware of the “Cautionary note” in the README (about not just using standard VCF-files).
Calculating likelihood ratios (’s)
No errors and matching genotypes:
LR_contribs <- wgsLR::calc_LRs_w(xT = c(0, 0),
xR = c(0, 0),
w = 0,
p = c(0.25, 0.25, 0.5))
prod(LR_contribs)
#> [1] 16No errors and non-matching genotypes:
LR_contribs <- wgsLR::calc_LRs_w(xT = c(1, 0),
xR = c(0, 0),
w = 0,
p = c(0.25, 0.25, 0.5))
prod(LR_contribs)
#> [1] 0Errors possible and matching genotypes:
LR_contribs <- wgsLR::calc_LRs_w(xT = c(0, 0),
xR = c(0, 0),
w = 0.001,
p = c(0.25, 0.25, 0.5))
prod(LR_contribs)
#> [1] 15.936Errors possible and non-matching genotypes:
LR_contribs <- wgsLR::calc_LRs_w(xT = c(1, 0),
xR = c(0, 0),
w = 0.001,
p = c(0.25, 0.25, 0.5))
prod(LR_contribs)
#> [1] 0.04761785Different error rates
Assume that the trace donor profile has and the suspect reference profile has . Then the is:
LR_contribs <- wgsLR::calc_LRs_wTwR(xT = c(1, 0),
xR = c(0, 0),
wT = 1e-4,
wR = 1e-8,
p = c(0.25, 0.25, 0.5))
prod(LR_contribs)
#> [1] 0.003198241Unknown trac sample error rate,
Assume that the trace donor profile has unknown but can be assumed to follow a beta prior on with mean value and variance :
mu <- 1e-4
sigmasq <- mu^2 / 2
shpT <-wgsLR::get_beta_parameters(mu = mu, sigmasq = sigmasq, a = 0, b = 0.5)
# 90% probability:
q <- wgsLR::qbeta05(c(0.05, 0.95), shape1 = shpT[1], shape2 = shpT[2])
q
#> [1] 1.776372e-05 2.371951e-04
curve(wgsLR::dbeta05(x, shpT[1], shpT[2]), from = 0, to = 1e-3, n = 1001)
abline(v = mu, lty = 2)
abline(v = q, lty = 3)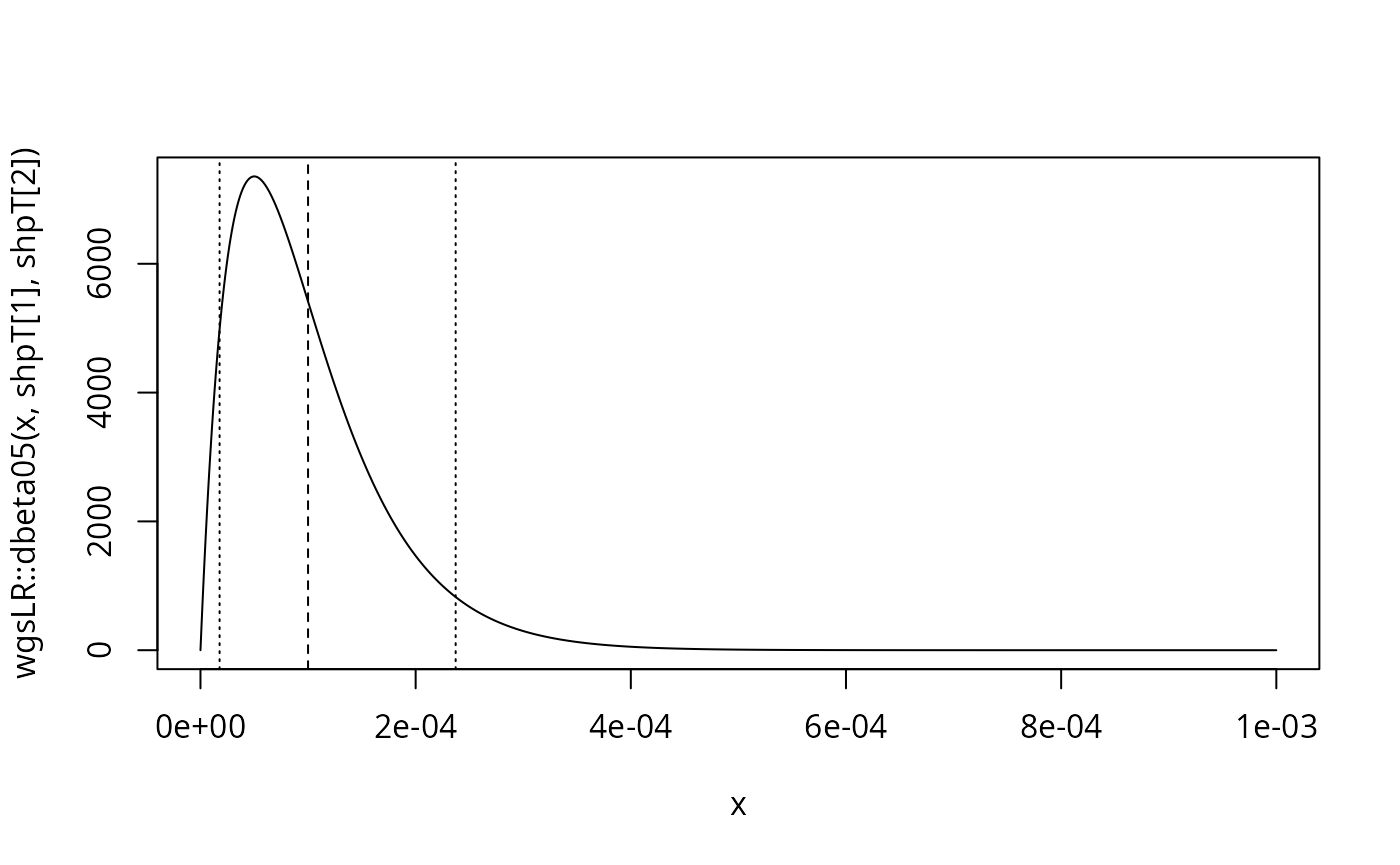
Further, assume that the suspect reference profile has . Then the is:
wgsLR::calc_WoE_wTwR_integrate_wT_mc(
xT = c(1, 0, 2, 2),
xR = c(0, 0, 2, 2),
shape1T_Hp = shpT[1], shape2T_Hp = shpT[2],
shape1T_Ha = shpT[1], shape2T_Ha = shpT[2],
wR = 1e-8,
p = c(0.25, 0.25, 0.5))
#> [1] -1.891458Verifying formulas
The usual
values,
for matches and
for non-matches, are obtained when setting
.
For example with both packages wgsLR and
caracas loaded we can do the following:
library(caracas)
LR <- d_LR_w$expr
LRw0 <- sapply(LR, \(x) as_sym(x) |> subs("w", 0) |> as_character())
with(d_LR_w, cbind(xT, xR, LRw0))
#> xT xR LRw0
#> [1,] "0" "0" "1/p_0"
#> [2,] "0" "1" "0"
#> [3,] "0" "2" "0"
#> [4,] "1" "0" "0"
#> [5,] "1" "1" "1/p_1"
#> [6,] "1" "2" "0"
#> [7,] "2" "0" "0"
#> [8,] "2" "1" "0"
#> [9,] "2" "2" "1/p_2"And with sample specific error rates, and :
library(caracas)
LR <- d_LR_wTwR$expr
LRw0 <- sapply(LR, \(x) as_sym(x) |>
subs("wT", 0) |>
subs("wR", 0) |> as_character())
with(d_LR_wTwR, cbind(xT, xR, LRw0))
#> xT xR LRw0
#> [1,] "0" "0" "1/p_0"
#> [2,] "0" "1" "0"
#> [3,] "0" "2" "0"
#> [4,] "1" "0" "0"
#> [5,] "1" "1" "1/p_1"
#> [6,] "1" "2" "0"
#> [7,] "2" "0" "0"
#> [8,] "2" "1" "0"
#> [9,] "2" "2" "1/p_2"